The need for AI in biosciences and medical applications is undeniable. The opportunities that arise thanks to the combination of pure computational power and human ingenuity are simply breathtaking. Thanks to AI, potentially unsolvable problems like the protein folding paradox have now been solved (Alpha Fold II). As analyzing the huge amount of data allow us to reach conclusions unavailable to humans, unimaginable advances like curing or even eradicating genetic diseases are now within our reach.
Before elaborating on the exact applications of AI techniques in genomics, let us introduce the basic concepts behind this sometimes mysterious artificial intelligence. Let’s focus on areas most commonly used in biosciences: machine learning and deep learning.
Machine learning is a subset of artificial intelligence. It is based on the algorithms that can learn from the data to improve their functioning in the future. It is done in the process called training when huge numbers of labelled data are used to create an algorithm with its own distinctive features that are consistent across all data. As a result, these algorithms are able to make predictions and decisions without explicit programming from humans.
The most common division of ML is:
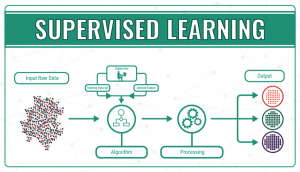
- Supervised learning, in which algorithms are trained using labelled examples, such as an input where the desired output is known. First of all, the data is split into a training set and a test set. Then, the algorithm is fit to the training dataset. The next step is to evaluate and adjust the model using test data, attempting to avoid overfitting, where the model’s predictive power is low, and underfitting, where the model doesn’t match the data properly. Supervised learning can be divided into classification problems, where a specific title or category is assigned to the input variable, and regression problems, when the output variable is a real or continuous value, such as height.
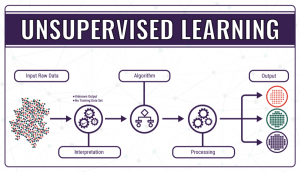
- Unsupervised learning, where the data do not have a categorical output or label assigned to them, so the ‘right answer’ is not known, and the model needs to figure out what it’s processing. The algorithm in unsupervised learning is trained to find trends, patterns or structures by studying and observing the unarranged data and creating its own connections between them. Learning and improving by trial and error is the key to unsupervised learning.
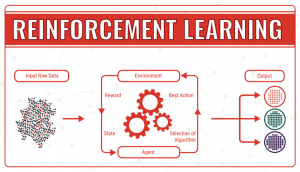
- Reinforcement learning, which operates thanks to the use of an algorithm that, through trial and error, identifies which particular input data provides the best numerical output data. It continuously tries to improve the outcome taking into account the feedback of the environment, which can either be a reward, which reinforces that the model achieves what was planned, or a failure, which prompts the model to change a strategy. In machine learning terminology, the environment is called a state, which can have different actions (responses). The goal is to continuously improve the quality of the outcome (Q). The algorithm needs to determine which actions improve the state, so the quality goes up. It continues endless simulations of actions and states until it finds the best strategy.
Deep learning is another subset of artificial intelligence. It is inspired by the activity of the parts of the human brain, such as neurons, to allow computer models to cluster data and make predictions with incredible accuracy.
In our contemporary society, most complex questions cannot be answered using single-layered machine learning algorithms. As recently the computational power has increased exponentially, it allowed the data to be processed in a more sophisticated way using deep learning.
Deep learning distinguishes itself from machine learning because it eliminates some of the data pre-processing that is typically involved with machine learning. Deep learning algorithms can process unstructured data, like text and images, and it automatically selects which features are most crucial for identification, eliminating the need for human intervention as the hierarchy of features does not need to be manually adjusted like in machine learning.
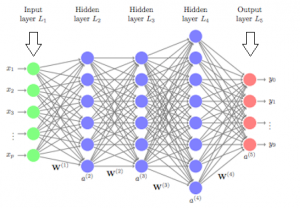
Deep neural networks are made of densely arranged layers of interconnected nodes, each building upon the previous layer to refine and optimize the outcome. This progression through the neural network is called forward propagation.
The input and output layers of a deep neural network are called visible layers. All the layers in between are called hidden layers. The input layer is where the algorithm starts to process the data. The information is then transferred through one hidden layer to another over connecting channels. Each has a value assigned to it, hence it’s called a weighted channel. All neurons have an associated number with it called bias. This bias is added to the weighted sum of inputs reaching the neuron, which is then applied to the activation function. The result of the activation function determines if the neuron is activated or not. Every activated neuron passes on the information to the next layer. This continues until the computations reach the output layer, where the final prediction or classification is made.
The weights and bias are continuously adjusted to produce a well-trained network that gradually becomes more and more accurate by self-correcting its errors. It happens by a process called backpropagation which uses algorithms like gradient descent to calculate errors and biases in predictions.
Now, moving to applications of AI in biosciences…
Genome annotation
When a genome is sequenced, it is not informative yet. It does not tell where the distinct elements of the genome are, which would be essential for almost any investigation in molecular genetics. The identification and annotation of these elements arose to address this task. This process is called genome annotation.
The genome can be interpreted as a huge amount of data that needs to be analyzed and categorized into subgroups. Databases have been analyzed by computer algorithms for a long time and this is the case with genome annotation as well. Algorithms can be trained to recognize patterns. In this case, the patterns are the different elements of the genome.
Transcription sites, splice sites, promoters, enhancers and positioned nucleosomes are all examples that were successfully analyzed and annotated thanks to these algorithms. In the following section, we will only focus on splice sites in order to provide a deeper understanding of such a process. It assumes the knowledge of basic statistical concepts such as conditional probabilities, Bayes theorem and the concept of sensitivity and specificity, so we strongly recommend our readers to make sure they understand these concepts.
Splice site prediction
Firstly, a tremendous amount of data was collected about the nucleotide sequences that occurred upstream or downstream of the splice sites for input for the machine learning algorithm.
These algorithms need features based on which they carry out their computations. In this case, the features were the presence or absence of the nucleotides very close to the splice sites.
The study used genes that corresponded to the splice site consensus, which suggests that there are GT sequences at the 5’ end of the introns and AG sequences at the 3’ ends of the introns.
Negative and positive instances were given based on this consensus: GT at the start of an intron and AG at the end of the intron are positive instances, while GT located within 100 nucleotides upstream the first GT and AG located within 100 nucleotides upstream the first AG are negative instances.
These instances had many features according to the following. One feature is the presence or absence of one nucleotide at a determined position. The study chose to observe 50 positions downstream and upstream the consensus. If we consider the combinations of the upstream and downstream positions as a feature, it gives us 400 features.
A data set was generated from many genes, using their information as input for the algorithm. These are called instances and are labelled with + if they are positive instances and with – if they are negative instances.
Our goal is to develop an algorithm that recognizes positive and negative instances that were not part of the training data set.
It is important to point out that there is not only one method that works, however, we only highlight one specific way to solve this problem here.
Classification method: The Naive Bayes Method:
This method classifies the instances based on the conditional probability that the instance is positive or negative given the features. If the conditional probability of being positive given a certain set of features is greater than the conditional probability of being negative given the same set of features, then the instance will be classified as positive and vice versa.
The conditional probability is computed using the Bayes theorem, hence the name of the method. With Bayes theorem, we can express the conditional probability of belonging to one class given the features in the function of the conditional probability of the features appearing in the class, the probability of the features, and the probability of the class. It is important to note that we consider the features to be independent of each other.
Feature subset selection:
The NBM is however not sufficient as it already builds on the relevant features that the algorithm does not know at this point. In machine learning, feature selection is a key step. Usually, a database provides many features which are not necessarily significant. If an algorithm does not eliminate an insignificant feature it will deteriorate the result. In this case, the algorithm must decide what nucleotides are important to take into consideration. When the features are selected, the algorithm can start carrying out the actual computation in order to classify the instance.
There are many feature selection methods known and they are suitable for different problems. One should always analyze the problem and the potential feature selection models to achieve the best result.
In our case, there are 400 features which means that there are 2400 subsets. Even a computer cannot randomly try all these subsets and see which one works the best. This is why we need so-called search algorithms. The performance is evaluated by computing the harmonic means of sensitivity and specificity. The sensitivity measures the ratio of the false negatives, the specificity gives the ratio of the false positives. The algorithm that the current study used is called the Backward Feature Elimination process. It starts with the subset which includes all features then analyzes features one by one and in every step eliminates the one that is the least relevant for the classification process. The determination of the least relevant happens through comparing the performances of the algorithm that includes a particular feature and the performance when the same feature is eliminated. Based on the effect that the elimination of one feature has on the performance it is possible to order the features according to relevance.
By continuously eliminating the features according to this order, it turned out that only the ten most relevant features made a significant difference. These features describe nucleotides near the splice sites.
Finding disease-related genes
It is well-known that social media platforms suggest people that you may know in order to add them as friends. These suggestions are made based on the common contacts that you and your already existing friends share. A similar method can be used by scientists to create maps of biological networks by analysing the interactions between different proteins or genes. The researchers from Linkoping University have shown in a new study that deep learning can be used to find disease-related genes. In AI, there are entities called “artificial neural networks” which are trained to find patterns in experimental data. These are currently used in image recognition, which can also be widely used in the field of biological research.
A remarkable example of how UCL contributes to this exciting AI development is the successful implementation of Eye2Gene. It is a decision support system to accelerate and improve the genetic diagnosis of inherited retinal disease by using AI on retinal scans. While there are over 300 possible genetic causes behind the disease, quick and accurate diagnosis dramatically increases the chances of successful treatment. It was designed by the research group led by Dr Nikolas Pontikos from the UCL Institute of Ophthalmology. Such few and pioneering examples of personalized medicine in practice significantly contribute to speeding up regulatory processes of various AI-driven technologies.
In other areas, like gene expression patterns, the scientists from Linkoping University used an enormous amount of experimental data: the expressions patterns of 20,000 genes from both people with diseases and healthy people. The information was not sorted before it was inputted into the artificial neural network; the researchers did not give information about which gene expression patterns were from healthy people and which were from the diseased group. The deep learning model was then trained to categorize the gene patterns.
One of the unresolved challenges of machine learning is not being able to see how the artificial neural networks solve their tasks and find the patterns of gene expression. We would only be able to see our input (the experimental data that we provide) and the output showing the result. In the end, the scientists were interested in which of the gene expression patterns found with the help of AI are actually associated with disease and which are not. It was confirmed that the AI model found relevant patterns that concur well with biological reality. At the same time, the model revealed new patterns that are potentially very important for the biological world.
Challenges and limitations of AI
The concept of artificial intelligence has gained a great level of interest in the last few years. AI is already deeply infiltrated into our everyday lives through, for example, our smartphones. However, by comparison, the use of AI in the healthcare system is not yet so advanced.
One of its limitations is that, even though people assume AI is objective and unprejudiced, biases can occur if they were already present in the dataset used for the input. One study showed that when texts written by humans in English were the initial database for an AI model, the algorithm made words associations similar with societal words associations found in the given texts, for example linking European names with more positive associations compared to some African names. Thus, these biases could raise serious issues in a healthcare setting for populations coming from different demographic areas.
One challenge for the further implementation of AI in healthcare is the threat it represents to the utility of human employees. The Topol Review anticipates that robots will be able to perform medical procedures without being controlled by humans as some robots already have a low level of AI controlling their physical actions. However, a study from 2017 reported that only 23% of people in the UK would be comfortable with robots performing medical procedures on them. Therefore, the AI models should not be developed to replace human employees but rather to supplement the work of the medical staff.
From a legal perspective, one other possible challenge would be the patients who seek legal action when an artificially intelligent machine fails to appropriately utilize genetic testing on them. Should the doctor who referred the patients be blamed or the programmer of the software? As mentioned before with the artificial neural networks, in AI, you cannot determine how the output of the programme was decided so this would only further complicate the answer to the question above.
Sources:
High Accuracy Protein Structure Prediction Using Deep Learning, John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Kathryn Tunyasuvunakool, Olaf Ronneberger, Russ Bates, Augustin Žídek, Alex Bridgland, Clemens Meyer, Simon A A Kohl, Anna Potapenko, Andrew J Ballard, Andrew Cowie, Bernardino Romera-Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reiman, Martin Steinegger, Michalina Pacholska, David Silver, Oriol Vinyals, Andrew W Senior, Koray Kavukcuoglu, Pushmeet Kohli, Demis Hassabis, In Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstract Book), 30 November – 4 December 2020.
https://datafloq.com/read/machine-learning-explained-understanding-learning/4478
Carreras, J; Hamoudi, R; Nakamura, N; (2020) Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. The Tokai Journal of Experimental and Clinical Medicine, 45 (1) pp. 37-48.
https://www.ibm.com/cloud/learn/deep-learning
https://www.ucl.ac.uk/ioo/news/2021/jun/eye2gene-wins-artificial-intelligence-health-and-care-award